The new EU clinical trial regulation (CTR) is set to revolutionise clinical trial processes across Europe impacting all European Member states and organisations wanting to run clinical trials across these regions.
How can organisations prepare for the change and set themselves up successfully with a new CTR programme?
Contents table
- What does the Clinical Trial Regulation hope to achieve?
- What are the significant changes you need to be aware of?
- About the CTIS: a new central portal for EU trials
- The new application assessment process
- New timings for trial notifications
- About the clinical trial publication
- What will the impact be for Sponsors and CROs?
- How can CROs like PHARMExcel support your clinical trial?
The long-delayed clinical trial EU Portal and Database, one of the main features of the Clinical Trial Regulation 536/2014 (CTR) and the key component of the Clinical Trial Information System (CTIS), is now finally fully operational from 31 January 2022.
1. What does the Clinical Trial Regulation hope to achieve?
The new regulation will bring about certain key changes to the clinical trial application process in the EU:
- Creating efficiency through harmonisation of the trial application process across Europe
- Providing greater transparency in clinical processes and data exchange
- Driving enhanced safety and efficacy of drugs.
In the words of the European Medicines Agency (EMA) the regulation’s aim is to create a landscape that is “favourable to conducting clinical trials in the EU, with the highest standards of safety for participants and increased transparency of trial information.”
The new regulation is applicable for Investigational Medicinal Products (IMP) for human use and does not apply to non-interventional trials or trials without medicinal products such as devices or surgery.
2. What are the significant changes you need to be aware of?
The CTR has brought key changes to the clinical trial application process across the EU.
Directive 2001/20/EC
- Implemented in 2004
- Multiple application submission for respective NCAs and ECs for each MSC for single trial
- Individual assessment, conclusion and decision for each MSC – No harmonisation
- Limited information available to the public
Clinical Trial Regulation 536/2014
- Published in 2014
- Harmonised process across EU-Single application submission to all MScs through single platform across the EU
- Coordinated assessment, conclusion and decision through reporting MSC
- All information (except agreed Personally Protected Data and Commercially Confidential Information) available to the public
3. About the CTIS: a new central portal for EU trials
CTIS is the centralised EU portal workspace and EU database which will be the single EU entry point for clinical trial applications. An application consisting of Part I and/or Part II will be created centrally, via the new CT Portal; Part I consists of information related to the trial, product and protocol whereas Part II consists of data specific to the member states where the trial will be run.
The CTR provides for three [1] different types of application dossiers;
- Initial application; the first application to be submitted by the sponsor providing comprehensive information about the trial to be conducted and the investigational medicinal product(s) to be used, enabling the authorities of the Member States Concerned (MSC) to evaluate the acceptability of conducting the trial- utilising new templates
- Substantial modification [2]; an application to submit a request for substantial changes to an authorised clinical trial
- Additional MSC application; An application to submit an additional member state to a previously authorised clinical trial- can be submitted when there are no other applications under evaluation.
[1] Note: A non-substantial modification is not considered an application as it is not subject to the evaluation and decision issued by the MSC
[2] A substantial modification is a change that either has a substantial impact on the safety or rights of the subjects; or on the reliability and robustness of the data generated in the CT.
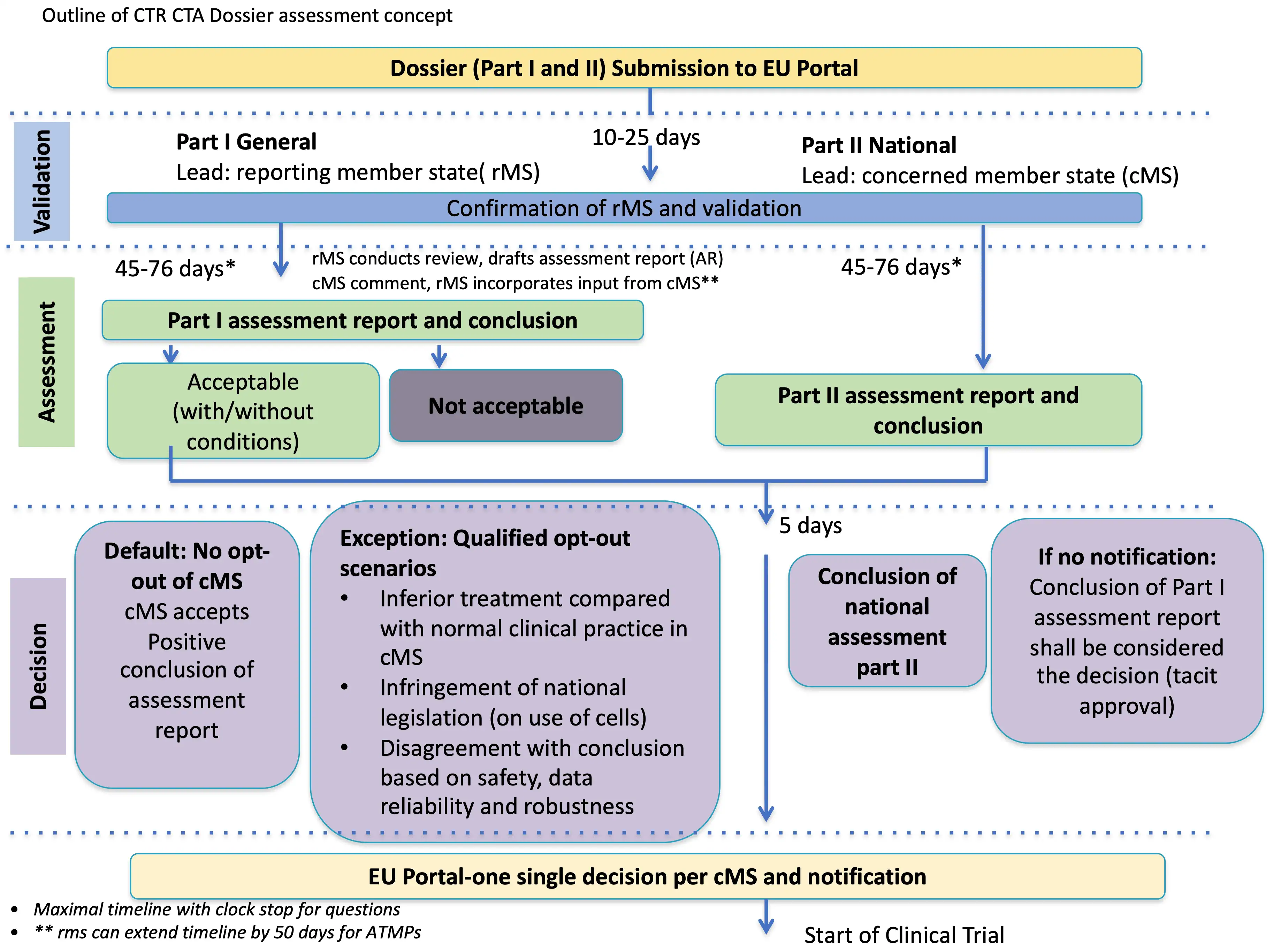
4. The new application assessment process
The assessment of Part I is carried out by the Reporting Member State (RMS) with the support of other MSC and the assessment of Part II is carried out by the MSC.
The initial application process
Throughout the assessments, sponsor organisations are expected to respond to information requests (RFI) raised by regulators within a 12 day timeframe.
5. New timings for trial notifications
Throughout the course of the clinical trial, sponsors are required to submit notifications within 15 days of the following main milestones;
- Start of trial
- First subject, first visit (FSFV)
- End of recruitment in a MSc
- End of trial in a MSc
- End of trial in all MSc
- End of trial in all third Countries Temporary halt
- Resuming after a temporary halt
- Early termination
6. Clinical trial publication
With the exception of sensitive and commercially confidential information, information stored in the database is published based on strict rules. Sponsors are allowed to manage the deferral of the publication via the portal.
7. What will the impact be for Sponsors and CROs?
Study start up; at the start of the study, some key sponsor operational processes such as site selection, product registration and application submission will be impacted.
During study conduct; project notification milestones must be submitted and tracked effectively to ensure all key milestones, including start of trial dates, subject recruitment dates and temporary halt dates, are submitted in a timely and validated fashion. Not submitting within the required timeline may have negative impact to the approval of trial.
Safety reporting; sponsors must ensure that issues, such as unexpected events, breaches and safety measures are submitted within the timeframe set in the regulation. These notifications are submitted via the portal and assessed by the respective member states.
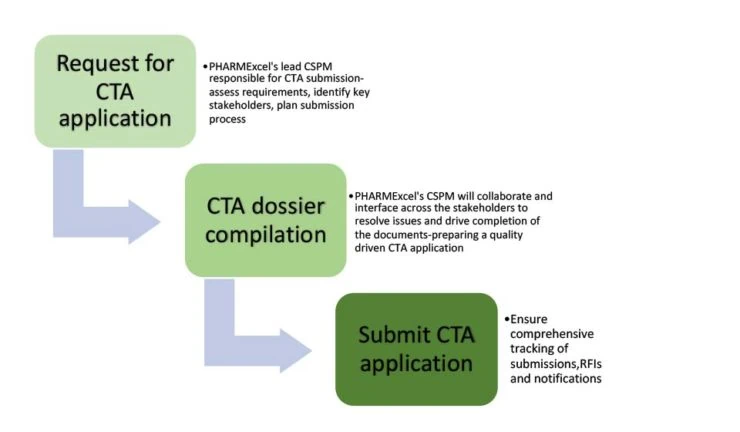
8. How can CROs like PHARMExcel support your clinical trial?
 Focus on first-time quality
Focus on first-time quality
Under the CTR, a CTAs success depends on the first-time quality of the dossier. A CTA will not pass the validation phase in any MSC unless it receives approval to proceed in all MSCs. The initial application authorisation procedure and any Substantial Modification application must be completed before you can submit an additional MSC. This greater emphasis on interrelationships across MSCs demands that sponsors get applications right the first time.
 Trial Experts-CTIS trained
Trial Experts-CTIS trained
PHARMExcel’s trial experts (all CTIS trained) can support the CTA process end to end, ensuring a “first-time right” CTA application dossier is compiled, whilst utilising comprehensive tracking systems to enable “real-time” reporting for sponsors.
 Local Knowledge
Local Knowledge
The study start-up strategy will need to change to a collective approach. Part I of the CTA dossier evaluation is a joint assessment from all MSCs led by the rMS. However, if a CTA contains elements or provisions at odds with the RMS’s national laws, a negative Part I conclusion will affect all MSCs.
 Focused team
Focused team
Communication entirely via CTIS and the short RFI response timelines require a focused team that monitors CTIS for incoming communications, handles document/data entry and download for trial master file compliance, and closely monitors timelines.
 Effective monitoring
Effective monitoring
Non-compliance with timelines may result in legal consequences for sponsors. PHARMExcel can diligently co-ordinate initial applications and all modifications. This will provide sponsors with efficiencies across all stakeholders and synchronisation of activities for each trial, as well as across all studies with the same product because an ongoing assessment in one MSC will block further substantial modifications to Part I, or Part I and II, for all MSCs.
PHARMExcel’s CTA process
We can support a smooth transition
From the 31st January 2022, the EU CTR is here to stay and once successfully in operation, will deliver many advantages to sponsors, assessors and the general public.
The PHARMExcel team can assist sponsors in smoothing the transition to the EU CTR by providing;
- CTIS trained, dedicated resource
- Quality driven application dossiers
- Focused oversight of all CTA applications and notifications.
We’re here to help, get in touch.