BACKGROUND
PHARMExcel was selected to manage a UK, Phase II, combined Investigational Medicinal Product (IMP)/Medical Device study for COVID-19 patients hospitalised in ICU.
We were asked to provide Project Management (PM), Monitoring and Safety oversight for the study, from protocol development to close-out. The study involved two sites and numerous teams responsible for key elements of the project. Our main goal was to bring everyone together, working fast and efficiently, ensuring adherence to regulations, whilst maintaining the highest quality standards.
ADDED VALUE
Our greatest strength throughout the whole process is having extensive experience of delivering on Investigator Initiated Studies (IIS). We were fortunate to already have a well-established relationship with the sponsor and were able to provide a highly experienced, dedicated PM and Clinical Research Associate (CRA) to work in collaboration with the teams.
1. Protocol, Investigator Brochure and Informed Consent documentation development
We provided expert and fast reviews of the key study documentation including the clinical study protocol, the Investigator’s Brochure (IB), and Informed Consent Documents (ICD). Our team’s experience of critical care studies, the combined product under investigation and the implementation of these types of studies, led to significant changes being made to these documents, ensuring they were accurate, concise and in line with applicable IMP and medical device regulations. These fast track reviews and changes allowed a smooth and fast approval pathway for the study.
2. Regulatory Approvals
The sponsor and clinical teams took responsibility for the Ethics/HRA and MHRA submissions with support from PHARMExcel. Our previous experience and understanding of combined IMP/Medical Device studies, (and gaining previous approvals) was invaluable to guide the sponsor through the application and submission processes.
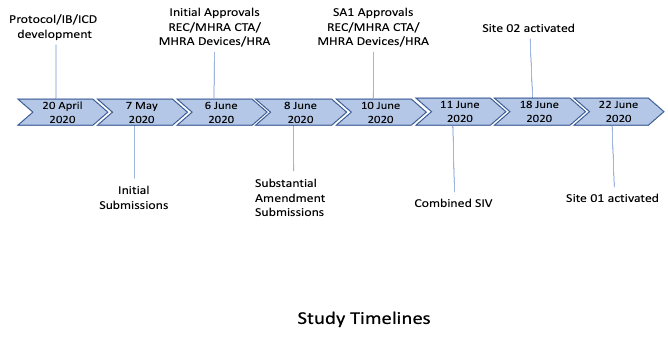
3. Study start-up procedures
Working with the sponsor and the wider study teams, our PM effectively and efficiently ensured the following project start-up/delivery elements were implemented:
- Efficient document management processes
- eCRF/data management development and testing
- Project tracking and risk management
- Development of key operational documents: IMP/Device Management Plan, Quality management plan, Communication Plan, Monitoring Plan
- Combined IMP/Device Safety Plan and adequate safety reporting/management procedures. Including the development of study specific SOPs and templates.
- Sample processing and management procedures
- Adequate patient management documentation

The rapid nature of the above was only achieved through daily communications between sponsor, manufacturer, study sites, pharmacy, laboratory and eCRF/Data Management vendors. All communications resulting in decision making and action items were recorded, and sponsor and manufacturer approvals sought where required.
4. Remote Site Initiation and Monitoring
PHARMExcel had worked extensively with both centres (although different clinical teams), so site feasibility was not required. Due to the current COVID situation and to ensure the study could be expedited, it was agreed a joint, remote SIV would be conducted. All key personnel from all parties attended the joint SIV and slides and recordings made available to all team members. The sites were activated as soon as the contractual process had been completed with each site. Expedited delivery of the combined product was undertaken by the manufacturers to ensure the sites had access as soon as they were activated.
The study CRA developed a risk-based Monitoring Plan (MP) that took into consideration the risks of the combined product, the patient population and inherent study procedures. The final plan was approved by the sponsor. Training and access to the electronic Case Report Form (eCRF) was undertaken to allow remote monitoring and site adaptations were made to enable remote Source Data Verification (SDV). A source data agreement was also established with both sites.
CONCLUSIONS
The above, whilst challenging due to the rapid set up of the study, provides a real-time example of effective Sponsor-CRO collaboration.
From initial study award, essential document development, through to combined approvals (including a further amendment), and initial site activation, took 8 weeks.
The sites have now commenced recruitment and we continue to oversee the running of the study to ensure regulatory compliance and recruitment targets are met.Author: Dr Filipa Martins (Head of Clinical Operations and Development)